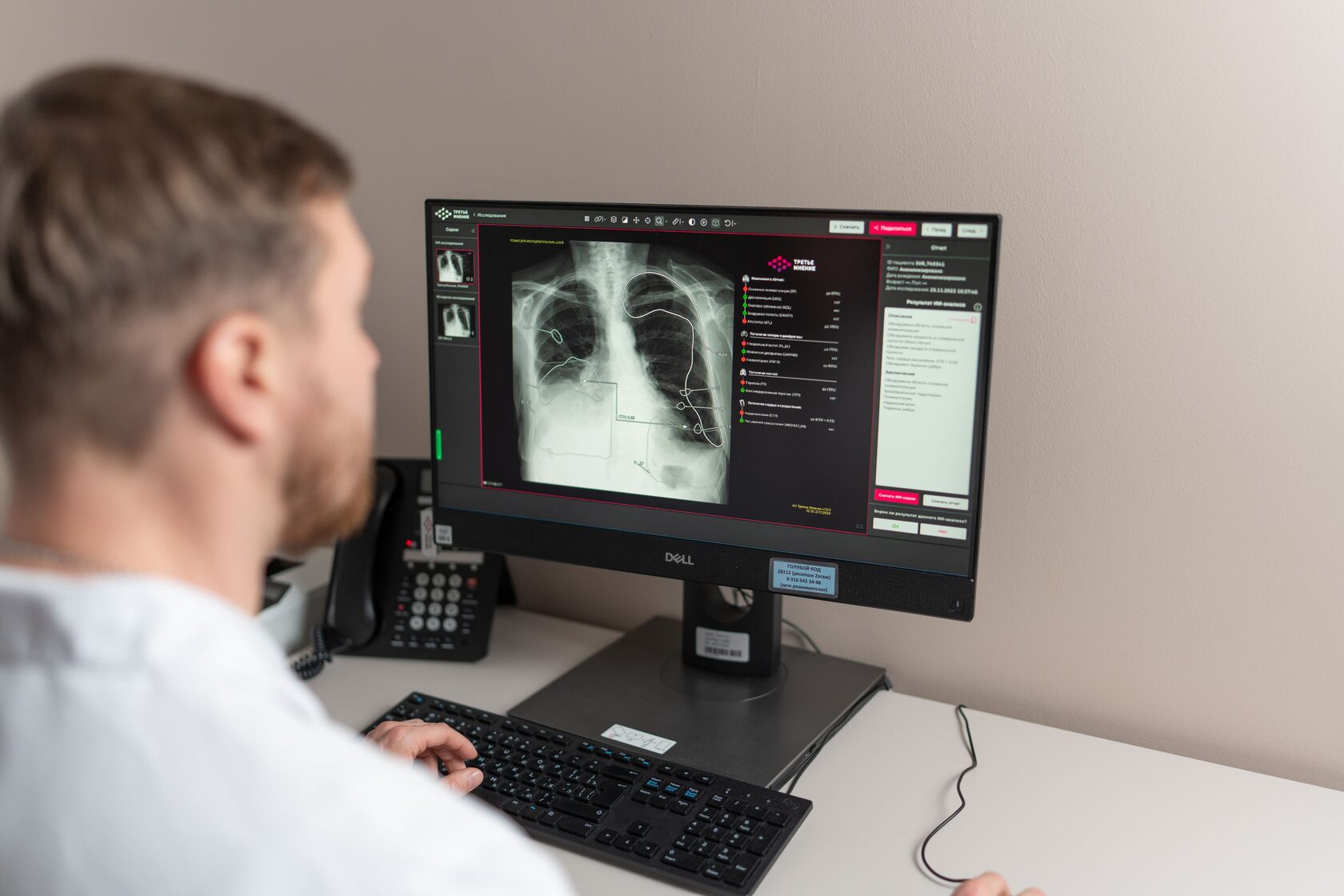
В 2025 FDA выпустило финальную версию руководства, как контролировать запланированные изменения в медизделиях с ИИ. Это переломный момент в нормативном регулировании медицинского ИИ.
Раньше разработчики медицинского ИИ сталкивались с проблемой: чтобы улучшить зарегистрированную версию продукта, нужно запустить повторную регистрацию. Это тормозило развитие и обновление сервисов.
В итоге несмотря на более высокие метрики, функциональность и пользу новой версии, продукт должен пройти долгую процедуру перерегистрации, которая по трудозатратам и времени сопоставима с первичной.
Теперь FDA разрешает компаниям заранее описывать и планировать обновления:
➡️ Какие изменения ждут алгоритм
➡️ Как они валидируются
➡️ Какие метрики безопасности нужны для подтверждения
Если план согласован — обновления можно внедрять без нового раунда «регуляторного цикла».
Почему это важно:
🟣 Для пользователей алгоритмы будут расти в эффективности и точности без потери безопасности и надёжности.
🟣 Для компаний открывается возможность делать ИИ-продукты «гибкими» и планово внедрять безопасные улучшения.
Все плановые изменения в ИИ-медизделии должны быть согласованы с системой менеджмента качества разработчика.
📌 В России компании-разработчики обязательно подтверждают соответствие стандарту ISO 13485-2017 «Изделия медицинские. Системы менеджмента качества».
Раньше разработчики медицинского ИИ сталкивались с проблемой: чтобы улучшить зарегистрированную версию продукта, нужно запустить повторную регистрацию. Это тормозило развитие и обновление сервисов.
В итоге несмотря на более высокие метрики, функциональность и пользу новой версии, продукт должен пройти долгую процедуру перерегистрации, которая по трудозатратам и времени сопоставима с первичной.
Теперь FDA разрешает компаниям заранее описывать и планировать обновления:
➡️ Какие изменения ждут алгоритм
➡️ Как они валидируются
➡️ Какие метрики безопасности нужны для подтверждения
Если план согласован — обновления можно внедрять без нового раунда «регуляторного цикла».
Почему это важно:
🟣 Для пользователей алгоритмы будут расти в эффективности и точности без потери безопасности и надёжности.
🟣 Для компаний открывается возможность делать ИИ-продукты «гибкими» и планово внедрять безопасные улучшения.
Все плановые изменения в ИИ-медизделии должны быть согласованы с системой менеджмента качества разработчика.
📌 В России компании-разработчики обязательно подтверждают соответствие стандарту ISO 13485-2017 «Изделия медицинские. Системы менеджмента качества».